Huntington disease
Huntington disease is an inherited disease that causes certain nerve cells in the brain to die. Dementia can result.

Overview
Huntington disease (sometimes shortened to HD) is an inherited disease that causes certain nerve cells in the brain to die. Huntington disease is one of more than 50 diseases or conditions that can cause dementia.
Symptoms of Huntington disease typically appear between the ages of 30 and 50. If a parent has Huntington disease, each of their children will have a 50 per cent chance of inheriting the gene mutation that can cause the disease.
As Huntington disease continues, a person's physical, emotional and cognitive functioning can be affected. A person may become less able to control movements, recall events, make decisions and manage emotions.
Other names
Other names historically used for Huntington disesase include:
- chorea
- chronic progressive chorea
- Huntington’s chorea
- hereditary chorea
The word “chorea” (a Greek term meaning "dance") is used to describe the involuntary movements that people with Huntington disease can experience.
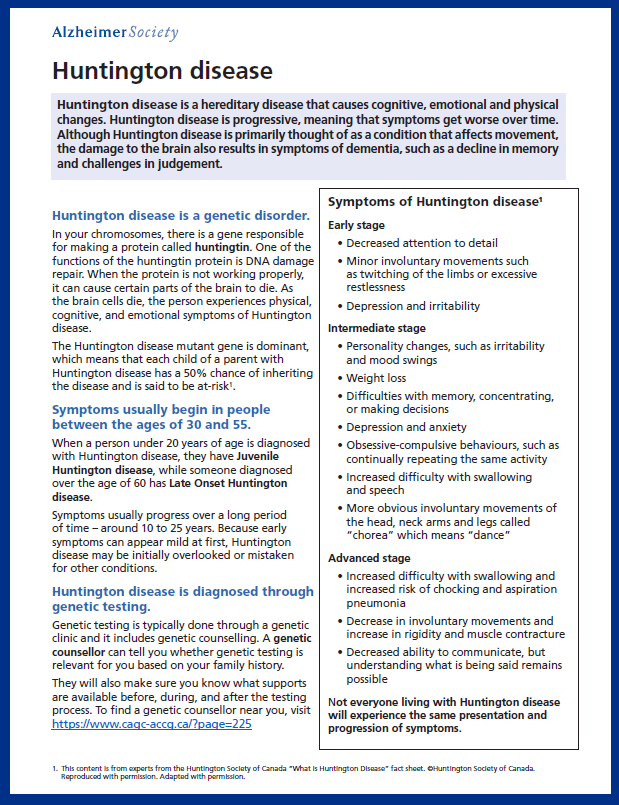
Symptoms
The age of symptom onset and the rate of the disease progression vary among people living with Huntington disease. However, symptoms typically begin in people between the ages of 30 and 50. The symptoms usually worsen over a 10- to 25-year period.
Changes in cognitive abilities and physical abilities
Early symptoms of Huntington disease can include subtle cognitive changes. Slight physical changes may also develop early on. For example, a person may experience an increased difficulty controlling their movements.
Changes in mood and personality
As the disease progresses, a person with HD may experience personality changes such as irritability, depression and mood swings.
Changes in judgment and memory
A person with Huntington disease may experience trouble with memory, concentration, learning new things or making decisions.
Obsessive-compulsive behaviour
Obsessive-compulsive behaviour, such as continually repeating the same activity, is also a common feature of Huntington disease.
Continued changes in physical abilities and cognitive abilities
Initial physical symptoms can gradually develop into more obvious involuntary movements such as jerking and twitching of the head, neck, arms and legs.
Later-stage symptoms of Huntington disease may include increased difficulty with walking, eating independently and swallowing.
Other symptoms at this stage may include an increased difficulty with concentration and communication.
Diagnosis
Medical history and exams
A thorough assessment for Huntington disease will often include:
- physical exams
- neurological exams
- psychiatric exams
- review of the person’s complete family medical history to help rule out other conditions
Changes in behaviour and personality, like increased irritability, can be mistaken for other conditions. This can cause some of the early symptoms of Huntington disease to be overlooked initially, and it's a reason why diagnosis examinations are so important.
Brain imaging, such as an MRI, is not usually needed. But it may be requested by a physician to detect any structural changes in the parts of the brain that are affected by Huntington disease or to rule out other conditions.
Genetic testing
People may undergo genetic testing via a blood test to confirm or rule out Huntington disease if they are exhibiting symptoms, such as challenges with thinking or moving.
Predictive genetic testing is also available for adults with a strong family history of Huntington disease. In this case, a result of "gene negative," "intermediate," "reduced penetrance" or "gene positive" is given. But a diagnosis of Huntington disease is not given until symptoms develop — usually much later in life.
Anyone interested in genetic testing should speak with a genetic counselor about risks and benefits of such testing. A genetic counsellor is also needed for help interpreting the results, which can be complex and depend on the number CAG repeats in the Huntington disease gene.
Typically, a physician or nurse-practitioner can refer a person for genetic counselling if they have a strong family history of an inherited disease.
Risk factors
Huntington disease is a familial disease. This means it can be passed from biological parent to biological child through a mutation in a gene. In this case, the mutation is in a gene responsible for huntingtin protein.
Anyone with a parent with Huntington disease has a 50 per cent chance of inheriting the gene. However, genetic results can be complex given that disease prediction is related to number of CAG repeats in the gene mutation, not just the presence of the gene itself.
In about one to three percent of cases, no history of the disease can be found in other family members.
Treatment
Medications can help manage some symptoms. But medications cannot slow down or stop Huntington disease. As a result, there is currently no cure for Huntington disease.
On a promising note, there are several drug trials underway. These may bring new treatment approaches.
Therapeutic approaches
Therapeutic approaches help people to manage symptoms.
- Occupational therapy can help improve the functional ability of people living with Huntington disease. This therapy can make use of assistive devices and other techniques.
- Physical therapy can help maintain physical abilities.
- Speech therapy can help improve verbal communication that may have been impaired by muscle-control issues. It can also help with eating and swallowing challenges.
Huntington disease can develop differently in different people, even within the same family or generation.
People living with Huntington disease, caregivers and professionals must work together to help manage the most effective treatment for each individual.
Video: Tim's real-life experience with Huntington disease
Tim explains he has been living with Huntington disease and young onset dementia his whole life – first, as the young child of a mother who had HD, and later, as a younger adult with symptoms himself.
In the years since his diagnosis at the age of 31, Tim has learned what helps him live well with Huntington disease and young onset dementia. This includes doing advocacy work with the Huntington Society of Canada, reducing other commitments and seeing his neurologist regularly. Find out more in this video produced in partnership by the Alzheimer Society of Canada and the Huntington Society of Canada..
More information and resources
Huntington Society of Canada. The Huntington Society of Canada offers ongoing support, education and information to help improve quality of life for Canadians impacted by Huntington disease.
For more information, read our print-friendly, downloadable brochure on Huntington disease in PDF.
This webpage was last updated on April 26, 2024.